发布日期:2020-01-15 13:56 浏览次数:
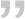
从各省市的药品GMP跟踪检查结果来看,涉及到压缩空气制备系统有关的问题层出不穷,究其原因在于各药企对于压缩空气制备系统的技术理解不够深入。

如河南省药品监督管理局2020年对药企的第一次飞检结果就表明,检查的65家企业其中就有6家企业发现8个压缩空气制备系统有关的问题需要整改。
就此,本文探讨了制药用压缩空气的质量要求和相应的制备技术,推荐了适用于不同剂型药品和不同洁净度级别的医药级洁净压缩空气质量标准和相应的制备技术。为制药用压缩空气的技术质量研究提供借鉴。
环境中每立方米空气约含上亿个尘埃微粒,大量细菌附着在微粒上,空气的相对湿度一般在45%以上。雾霾天气时空气中的污染物更多。空气在压缩和输送过程中不可避免地与机器部件接触,因此空气压缩机输出的压缩空气中含有以下杂质:
① 固体微粒:空气压缩机吸气过滤器无法消除空气中的微粒,空气压缩后固体微粒会增加;
② 水:空气经压缩冷凝后,即成为湿饱和空气,会夹带大量的液态水滴,即使是经分离的纯饱和空气,随着温度的降低,仍会有冷凝水析出;
③ 油:高速、高温运转的空气压缩机采用润滑油以起到润滑和密封作用,但会污染压缩空气;
④ 微生物及部分有害的化学异味物质。

① 压缩空气中的微生物粒子会污染产品;
② 混合在压缩空气中的油蒸汽聚集到一定程度就会形成易爆易燃源,润滑油汽化后会形成一种有机酸,易腐蚀压缩空气管道内表面;
③ 混入的微小颗粒极易损坏气动元件,更严重的是极易对物料造成污染;
④ 混合在压缩空气中的水分,在一定的温度压力下就会饱和析出水滴,当压缩空气与物料接触时,极易对物料的质量造成严重的影响。这些杂质会对生产过程和产品质量造成影响,甚至造成产品变质,导致经济损失甚至严重后果。
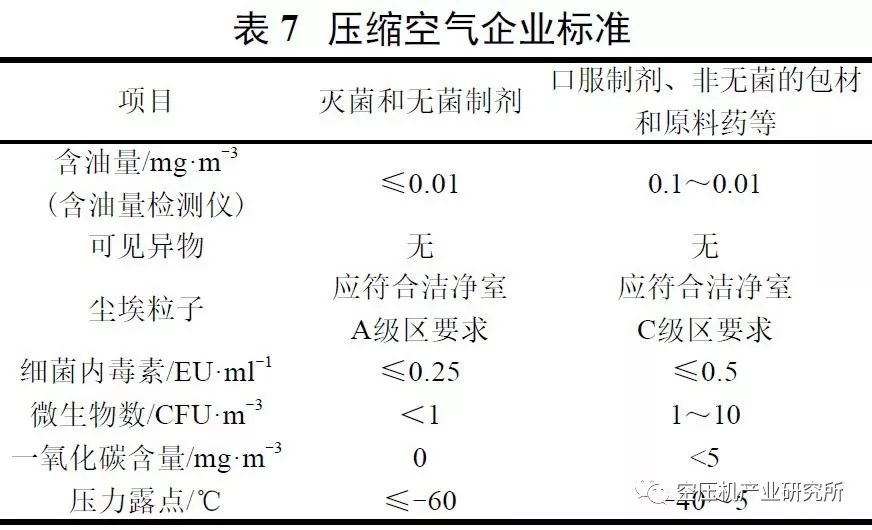
为保证药品质量,应控制压缩空气中的微粒数、含水量、含油量和微生物数等技术指标。
① 压缩空气中微生物数较高,检测合格率低;
② 含油量较高,难以达到医药级要求;
③ 空气压缩系统压力不稳定,影响药品生产质量。
1.1
压缩空气主要用途
在药品生产企业中,经常用到的压缩气体有空气、氮气和二氧化碳。
其中压缩空气的应用最广泛,作为工艺源气方面:
▶ 喷雾干燥装置及一步制粒机的喷液等工艺过程。
1.2
压缩空气杂质对药品的危害
在作为工艺源气使用时,压缩空气直接与物料和药品接触。在作为动力源使用时,虽不直接接触药品,但大部分是在洁净区内使用。

因此,制药用压缩空气必须使用洁净压缩空气,药品生产企业主要是要控制压缩空气的含水量、含油量、含尘粒量和含生物粒子量,同时还要求压缩空气无异味。含油压缩空气直接与药物接触会污染药品而滋生细菌。含水会加速细菌的生长和药品吸潮变质。
依据药品管理法要求,药品生产必须按照药品生产质量管理规范(GMP) 组织生产。GMP要求对进入洁净区的所有材料进行净化。因此制药用压缩空气必须按GMP要求进行管理和控制,才能保证药品质量安全。
1.3
压缩空气质量等级标准
压缩空气质量等级标准ISO8573.1 把压缩空气中的污染物分为固体杂质、水和油3种。我国等同采用了该标准。具体见表1。
1.4
制药用压缩空气质量标准
目前,制药用的压缩空气还没有国际和国家法定质量标准。《药品生产质量管理规范(2010年修订)》第八章第四十二条规定,进入无菌生产区的生产用气体(如压缩空气、氮气,但不包括可燃性气体) 均应经过除菌过滤,应当定期检查除菌过滤器和呼吸过滤器的完整性。

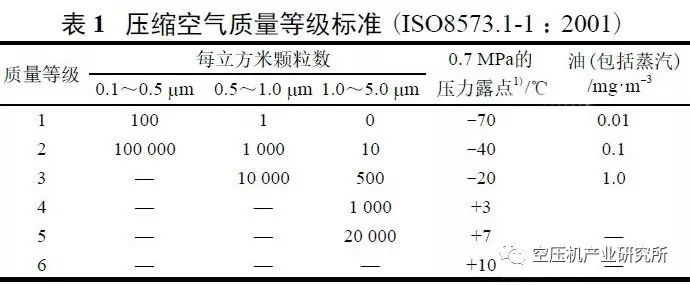
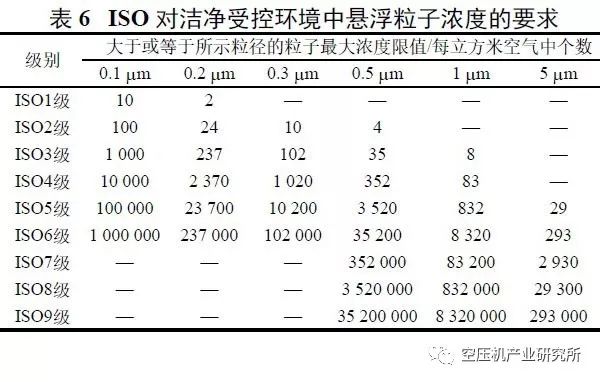
GMP无菌药品附录第九条规定:无菌药品生产所需的洁净区可分为4个级别。
以上各级别空气悬浮粒子的标准规定见表3。
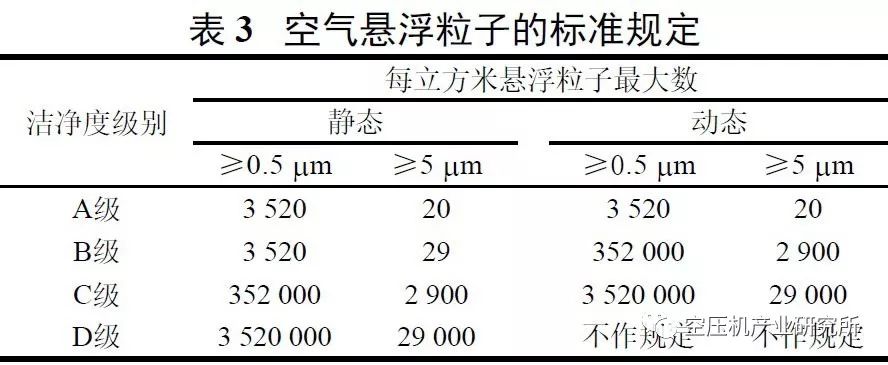
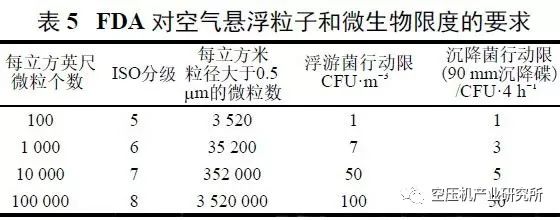
根据国内外药品相关法规和多年实践,压缩空气的品质可用4项指标衡量。
① 含水量用压力露点表示;
② 含尘量用尘埃粒径和单位体积内的数量表示;
③ 含油量用单位体积内含油质量表示;
④ 生物指标用单位体积内微生物数和细菌内毒素表示。
⑤ 无色,无味;不得含有正常空气组成之外的气体。一氧化碳是压缩空气中最危险的污染物,空气压缩机本身运转过程产生的排烟是其主要来源。因此,控制一氧化碳的含量<5mg/m³。

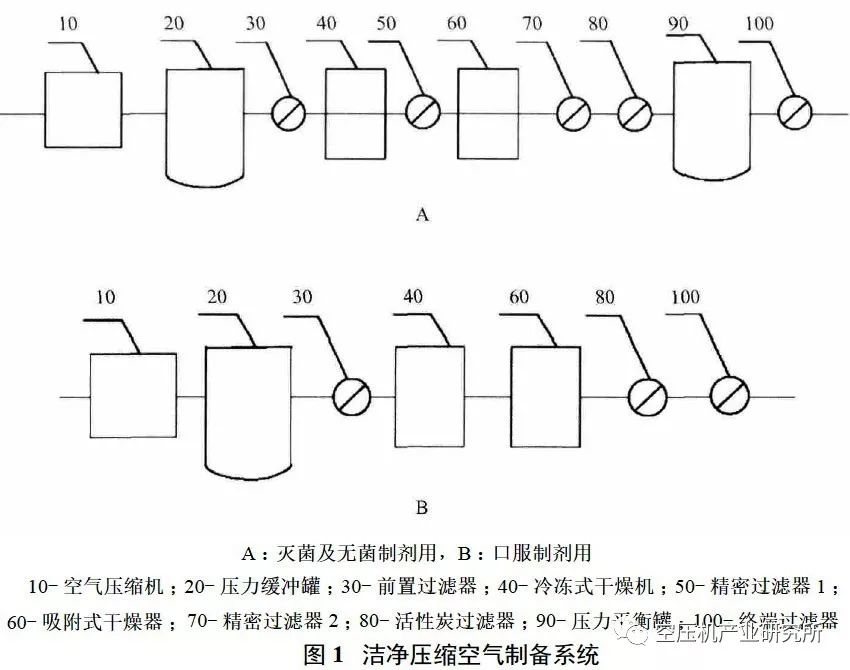
① 产生压缩空气部分:包括空气压缩机及其控制系统、冷却系统;
② 处理压缩空气部分:包括干燥机、过滤器;
③ 输送压缩空气部分:包括储气罐、管路、阀门。
3.1
洁净压缩空气制备系统工艺流程的选择
上述工艺流程根据工厂规模、空气压缩机类型及对压缩空气品质要求等的不同而有不同的配置,设计时应详细研究和分析用户的用气指标要求及用气特点,然后制定出合理的压缩空气净化工艺系统,选择确定合适的工艺设备。
3.2
空气压缩机及干燥过滤设施的选择
常用的空气压缩机有活塞式、螺杆式和离心式3种。离心式空气压缩机单台容量大,在小气量时易发生喘振,故该机型对药品制造企业不适用。活塞式空气压缩机结构尺寸大,易损部件多,噪声和振动也较大,且自动化水平较低,故药品制造企业也很少采用。螺杆式空气压缩机结构紧凑,占地小,仅需轻型基础,自控水平高,所以较多采用。
螺杆式空气压缩机又分为无油型和喷油型两种。在制药工程设计压缩空气站时,应首选无油螺杆式空气压缩机。近年来,随着压缩空气后处理除油技术的发展,喷油螺杆空气压缩机配备精密高效除油器,产出的气体含油量可达0.1mg/m³以下,基本上能满足口服制剂制药用的要求。但无菌和灭菌制剂还须选无油型空气压缩机,以确保其产品质量。
3.2.2 干燥装置的选择
压缩空气的干燥方式,分为冷冻式和吸附式两种。在压缩空气的压力露点要求大于3 ℃时,通常采用冷冻式干燥机。这种干燥装置具有流量范围大、供气连续、压力稳定且无压缩空气消耗等特点。在压力露点小于3 ℃时,则采用吸附式干燥机或冷冻式加吸附式组合干燥装置。
3.2.3 过滤器的选择
3.3
洁净压缩空气制备系统设计的技术问题
① 缓冲罐的设置和选择。在大中型压缩空气系统中用气点多,各用气点的用气量不同,如遇用气量高峰,系统压力波动很大。压力过低,影响使用,压力波动会造成过滤器损坏,影响压缩空气的洁净度。此时就要首先考虑在空气压缩机后、冷冻式干燥机前设置储气罐。如果要求压力波动小、洁净度要求高,则需在用气点洁净区内再设置一个带呼吸器的不锈钢平衡罐。
② 工艺源气和仪表用气通常共同建设于厂房内的一般区,合建1个空气站。系统供气压力按最高用气压力确定,较低用气点的压力通过在终端过滤器之前设减压阀来实现。
③ 输送管网所用阀门和管路材质均采用不锈钢材料,以316L型不锈钢材质为最佳,管路焊接要用氩弧焊。防止系统腐蚀。
④ 在工厂空气压缩站设计中,通常采用风冷式螺杆压缩机室内吸气,此时要加强室内的通风。一是降低温度,二可防止空气压缩机产生的一氧化碳的吸入。
⑤ 在工厂空气压缩站设计中,通常采用风冷式螺杆压缩机室内吸气,此时要加强室内的通风。一是降低温度,二可防止空气压缩机产生的一氧化碳的吸入。
⑥ 采用冷冻式干燥机时,前置过滤器应设置在其上游。
⑦ 为保证最终洁净度,用气终端需用0.22μm过滤器过滤;必要时则须用0.1μm 过滤器。
4.1
举例医药级洁净压缩空气的制备装置及方法
对实例1~4所制备的医药级洁净压缩空气进行检测。
4.2
检测项目、方法及结果
① 含油量测试方法:在压缩空气终端用气点,用压缩空气质量检测仪专用采样管检测。
② 可见异物和细菌内毒素取样及测试方法。在压缩空气终端用气点,将压缩空气注入密封瓶内,保持一定压力,让气体从瓶底部缓缓通过灭菌注射用水,用专用采样器设定流量数,当通气量达到设定值时,取出采样器,包好密封瓶,备检;
③ 取上述供试液,采用中国药典2015年版规定的细菌内毒素检查法检查;
④ 尘埃粒子取样及测试方法。在终端用气点,取洁净的试验用5L锥形瓶,锥形瓶直立,将压缩空气调到合适压力,通入瓶中,排空全部空气。将尘埃粒子计数器采样口置于锥形瓶中下部,检测尘埃粒子数;
⑤ 微生物数测试方法。在压缩空气终端用气点,通过减压阀与压缩空气取样装置连接,并将灭菌后的采样装置与浮游菌采样仪连接,按照浮游菌检测规程检查;
⑥ 压力露点测试方法。在压缩空气终端用气点,用压缩空气露点仪检测。
上述项目测试结果见表8:
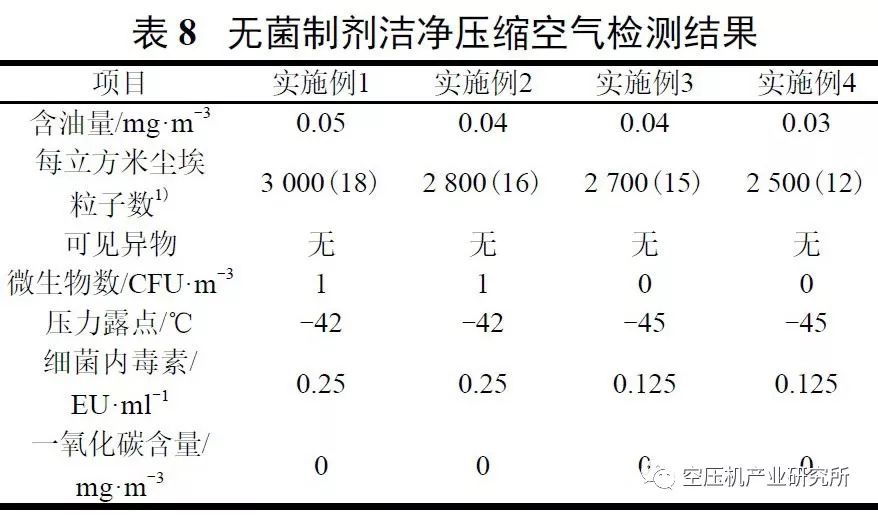
由表8中数据可知,采用图1A所示医药级洁净压缩空气的制备装置及方法制得的压缩空气可用于直接接触药品或药品的内包装材料生产,能保证产品质量。
4.3
结语
在药品生产过程中应根据不同产品的质量要求,对所使用的压缩空气进行洁净处理。通过先后设置冷冻式干燥机和吸附式干燥器,同时通过依次设置前置过滤器、精密过滤器、活性炭过滤器和终端过滤器的过滤组合方式,充分除去空气中的尘埃粒子、油蒸汽及微生物,确保获得技术指标符合表7中要求、适用于不同剂型和药品特性、洁净度级别不同的制药用洁净压缩空气。
药品安全是不容忽视的重大民生问题。药品制造行业所用压缩空气对药品质量有极其重要的作用,因此控制压缩空气的质量至关重要。对于压缩空气系统设计、施工和质量标准及应用管理等诸方面存在的问题,应引起有关企业的高度重视。
转载:空压机产业研究所
仅供大家参考学习使用,如有侵权,请联系删除
